James Foadi(a) and Pierre Aller(b)
(a) Imperial College London (j.foadi@imperial.ac.uk) , Diamond Light Source Ltd (james.foadi@diamond.ac.uk)
(b) Diamond Light Source Ltd (pierre.aller@diamond.ac.uk
In this tutorials it will be shown how to use the program BLEND to create a complete data set out of many small sweeps collected from 56 crystals at room temperature, directly from crystallization plates. References, documentation and more tutorials on BLEND can be found browsing the CCP4 website (http://www.ccp4.ac.uk).
The structure is an integral membrane protein called Tellurite resistance protein TehA homolog from the hemophilus influenzae organism. This structure has been solved initially to 1.2 Å resolution from a single, cryocooled crystal (Chen et al. (2010)). Deposited data can be downloaded (code 3M73) and used to explore data quality of the assembled data sets. Data and some of the methods presented in this tutorial have been previously submitted as part of a publication (Axford et al. (2015)).
We will assume for convenience that "blend_tutorial" is the name of a directory created in the $HOME area ("/home/vtv54516" in our case), or any other suitable directory, and in which all data related to this tutorial will be created. Different directory names and locations can be used, but appropriate modifications will have to be applied to the paths displayed in this document.
The above link allows to download the file "dataTehA.tgz". Copy or move this file in the "blend_tutorial" directory. Then extract the data with the following command line (assuming you are in "blend_tutorial"):
tar -zxvf dataTehA.tgz
The command line unpacks a directory named "dataTehA"; 67 data sets are included in this directory. These are "MTZ" files resulting from the conversion of XDS "INTEGRATE.HKL" files with POINTLESS. "MTZ", rather than "HKL" files are used in this tutorial to speed up tutorial execution, but "HKL" files can be used directly in BLEND and the conversion managed internally.
In this tutorial BLEND will be executed using command-line syntax. Equivalent execution can be obtained using the CCP4 GUI, ccp4i.
Before starting this tutorial you should already be in the directory "blend_tutorial" and should have all data in the directory "dataTehA". If this is not the case, please read the previous section "DATA PREPARATION".
Prepare the environment for CCP4 and R according to whatever set of instructions is applicable to your system. Next, in order to number all 67 data sets with the same serial numbers shown on this document, use file "original.dat" in directory "dataTehA". Simply copy this file in "blend_tutorial":
cp ./dataTehA/original.dat .
Now we are ready to execute BLEND in analysis mode, to analyse all 67 data sets and calculate the clustering dendrogram. Just type the following command line:
blend -a original.dat
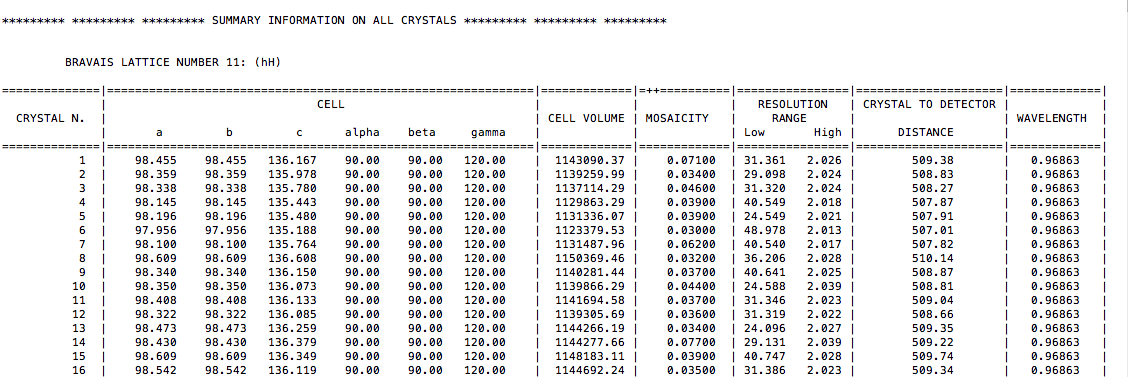
After dumping a few lines the programs awaits input keywords; for now simply press the "ENTER" key and wait till execution terminates. A number of files has been produced. A quick summary of all data sets read in BLEND is provided by "BLEND_SUMMARY.txt", the top part of which looks like this:
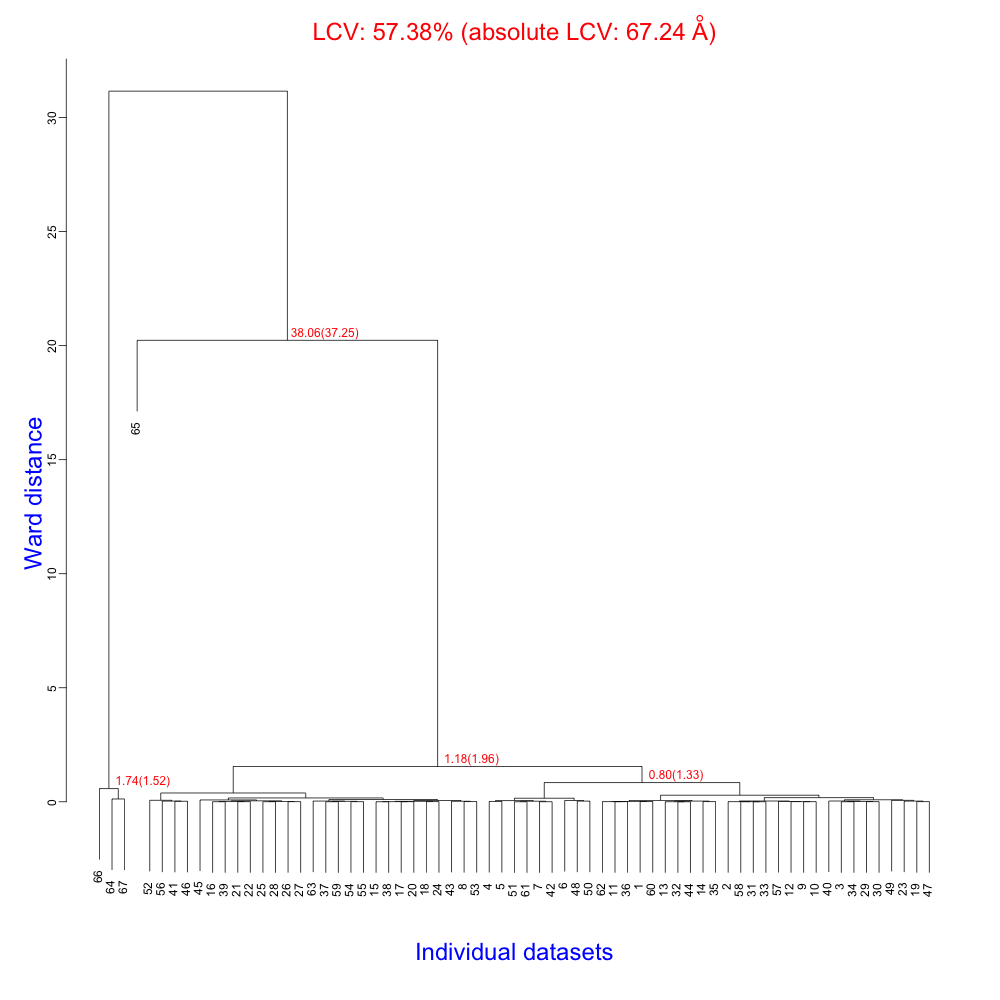
Clustering is represented by the dendrogram in the PNG file "tree.png":
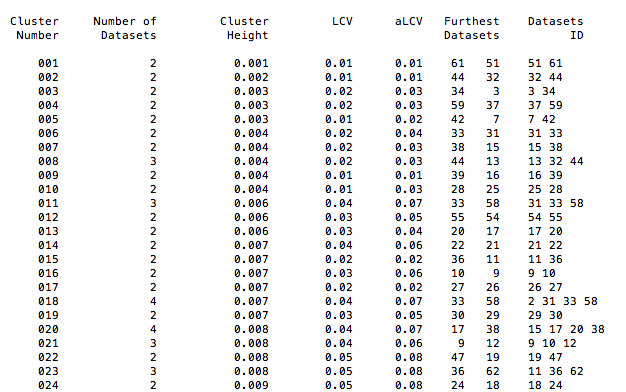
The ASCII version of this dendrogram is given in file "CLUSTERS.dat", whose top part looks like the following:
The linear cell variation (LCV) and absolute linear cell variation (aLCV) indicators show very large values when data sets 64, 65, 66 and 67 are involved. Normally this indicates some sort of failure in data indexing or that the structure has crystallized in a different form. Given that these four sweeps are a very tiny fraction of all data, we are much better off getting rid of them. In order to do so, let us create a new directory, called "second_run":
mkdir second_run
Next copy file "mtz_names.dat" in directory "second_run" as "original.dat" and let's set ourself in that directory:
cp mtz_names.dat ./second_run/original.dat
cd ./second_run
Now we need to edit "original.dat" in order to delete the last four lines, as they correspond to data sets 64, 65, 66 and 67. Eventually BLEND can be executed again in analysis mode like before. The new dendrogram looks like this:
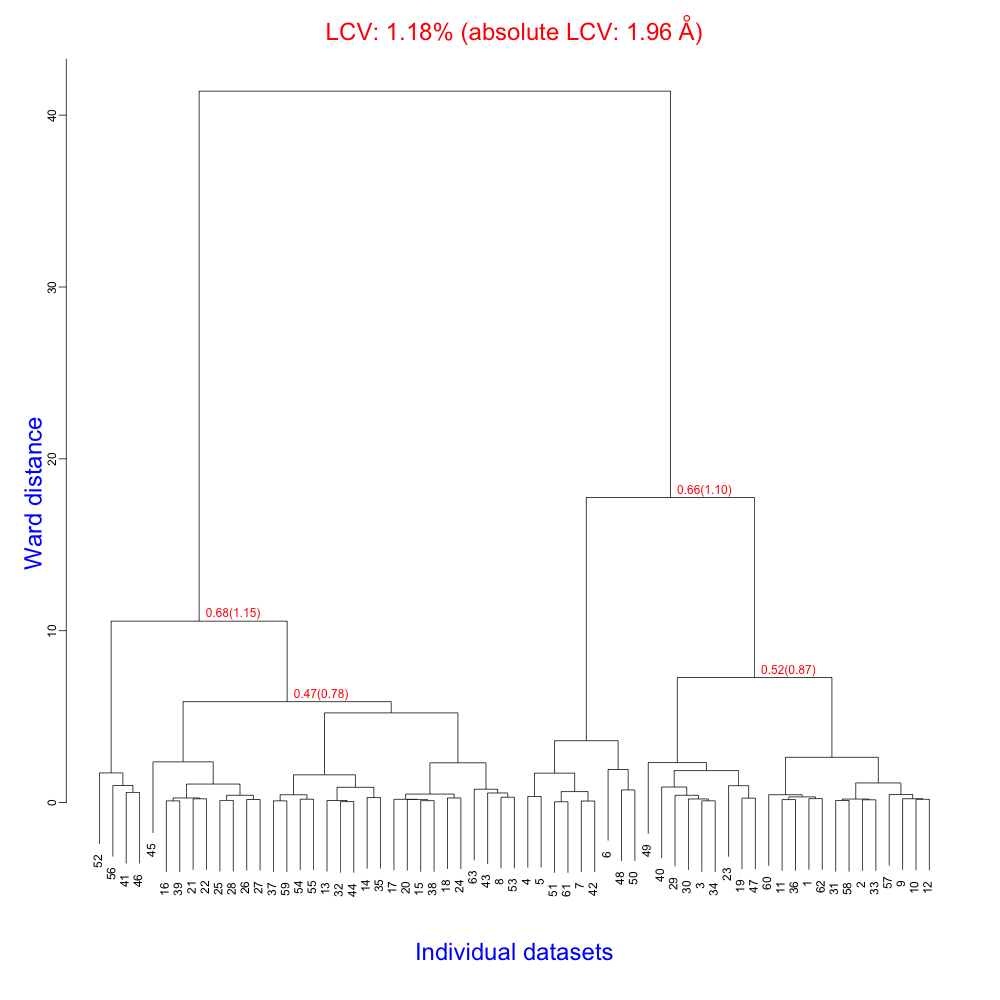
LCV and aLCV values have slightly changed because some of the clusters have also changed slightly. But the range is within acceptable values, as far as isomorphism is concerned. The most noticeable feature of this dendrogram is that it is split into two branches having, more or less, the same number of individual data sets. We can, therefore, proceed to run BLEND in synthesis mode, limiting resolution to an acceptable value. This can be inferred from the file "FINAL_list_of_datasets.dat", the top part of which is displayed here:
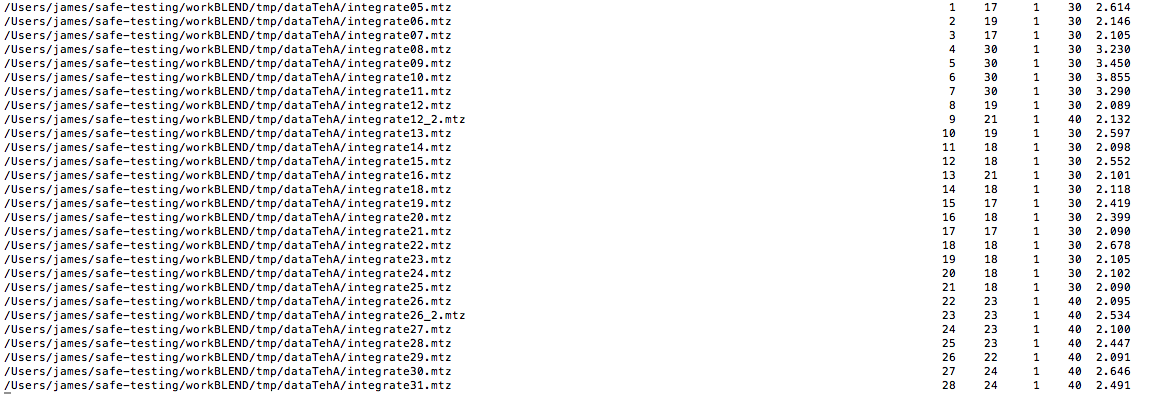
The first column contains full path to all input data files; the second contains the serial number assigned to input files; the fourth and fifth columns list initial and final image numbers, while the third lists the last image BLEND will include for all subsequent scaling and merging jobs. Numbers in these columns have been calculated through procedures to get rid of radiation damaged images. The last column includes resolution cuts suggested by BLEND for all subsequent scaling and merging jobs. These are worked out using intensity averages decay with resolution, and are controlled by keyword ISIGI. Here there are several data sets with suggested cut at around 2.1 Å and some others at higher or lower resolutions. As the algorithm for resolution cutting in BLEND (a polynomial interpolation of intensity averages) is quite conservative, we can attempt at fixing resolution to 2.1 Å for all synthesis and combination jobs.
To run BLEND in synthesis mode and have scaled and merged data corresponding to each node of the dendrogram we select just one value; in this case it will have to be higher than the highest node value. This can be read in file "CLUSTERS.dat". It is 41.395 and corresponds to cluster 63. Thus the single value to be used in the synthesis run can be 42, because all node heights are less than 42. Also, we need to prepare a keywords file to be fed to the program so that we don't have to input values manually. Thus, let's prepare an ASCII file called "bkeys.dat" with the following lines (find out the meaning of TOLERANCE by looking at BLEND documentation):
TOLERANCE 100
RESO HIGH 2.1
Then BLEND can be executed in synthesis mode with the following command line:
blend -s 42 < bkeys.dat
A new directory, called "merged_files" has been created. In it BLEND has stored all files involved in the scaling and merging process related to all nodes of the dendrogram. A very useful file is "MERGING_STATISTICS.info", which collects overall statistics for all selected node. The top part of this file is here shown:
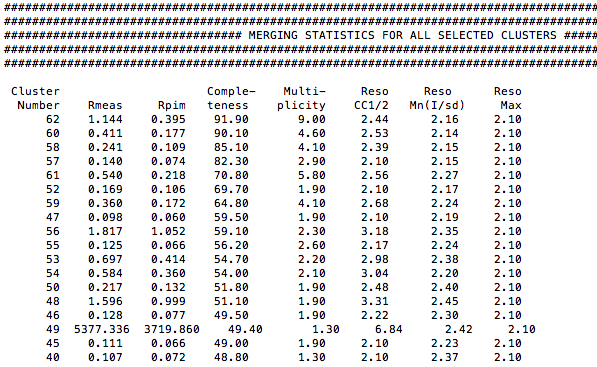
Clusters 60 and 61 are associated with the two main branches of the dendrogram. The crystals composing them have a good degree of isomorphism because the aLCV values for them are 1.15 Å and 1.10 Å, respectively. But the completeness for cluster 61 is quite low. Therefore we will have to use cluster 60 to assemble a complete data set.
Statistics for cluster 60 can be improved if we filter out some sweeps. Hopefully resolution and overall quality of the resulting data set will improve accordingly. Aim of this tutorial is to work out which sweeps need to be eliminated by executing BLEND in combination mode repeatedly.
Hint: data sets 45 and 46 seem to be the problem here. Start by creating the plot shown in the following picture (use BLEND graphics mode for this):
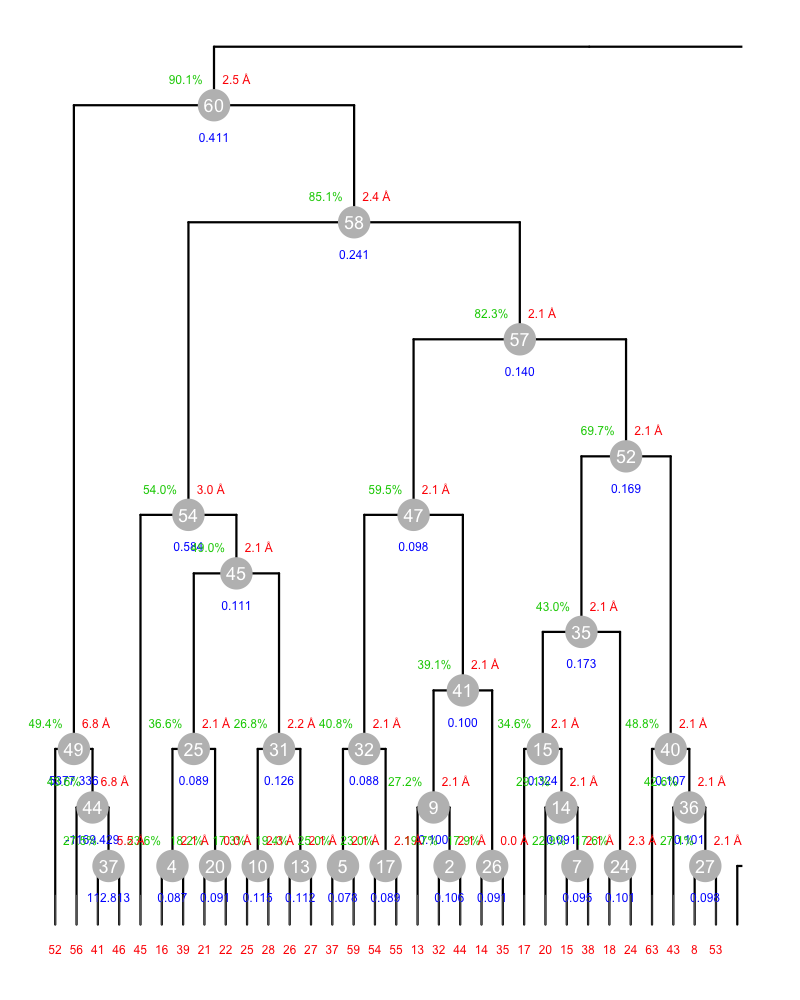
Although this annotated dendrogram looks cluttered because too many nodes are displayed, you should be able to see that all clusters at the bottom have reasonable values of Rmeas, but cluster 49. How can cluster 49 be improved? The same goes for clusters 54 and 58. Eventually, will the improvements on these clusters have effect on cluster 60?
(2010)
Chen, Y.H., Hu, L., Punta, M., Bruni, R., Hillerich, B., Kloss, B., Rost, B., Love, J., Siegelbaum, S.A., and Hendrickson, W.A.,
"Homologue structure of the SLAC1 anion channel for closing stomata in leaves", Nature 467, 1074-1080
(2015)
Axford, D., Hu, N-J., Foadi, J., Choudhury, H. G., Iwata, S., Beis, K., Evans, G. and Alguel, Y.,
"Structure determination of an integral membrane protein at room temperature from crystals in situ", (submitted)